
D’où vient le SARS-CoV-2 ? Cette question aurait pu aussi se poser pour le SARS de 2003 qui fut éradiqué parce que seuls les patients atteints du syndrome respiratoire étaient contagieux. J’ai fait ma propre enquête en privilégiant certains caractères de ce virus. Quelques-unes des analyses présentées sont inédites et ne figurent pas dans des publications scientifiques.
1) Les coronavirus
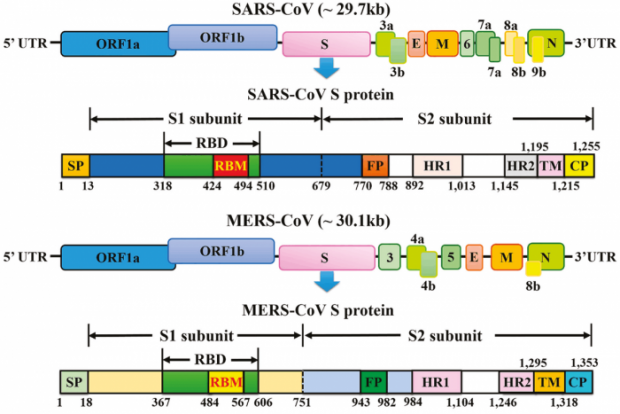
Les coronavirus sont des particules composée d’un génome à ARN de quelque 30 000 bases, associé à une enveloppe virale composée de trois types protéiques, M pour membrane, E pour enveloppe et S pour spicule. L’ARN est associé à la protéine N et forme une nucléocapside. Cette protéine N joue un rôle important lorsque le génome entre dans une cellule. A ces quatre protéines dites structurales s’ajoutent 16 protéines non structurales, codées sur les deux premiers tiers du génome (ORF1a,b), parmi lesquelles la nsp12 est une polymérase permettant de répliquer le génome. Pour finir, il y a aussi des protéines dites accessoires, codées sur des séquences de lecture vers la fin du génome. Les coronavirus se scindent en quatre types, alpha, bêta, delta, gamma. Les deux premiers auraient comme réservoir d’origine la chauve-souris ; ils infectent une série de mammifères ainsi que les humains. Les types delta et gamma auraient comme origine les oiseaux ; ils infectent en plus les bovins. Ces coronavirus sont en fait endémiques, présents dans un réservoir comprenant une ou plusieurs espèces. La distinction endémique épidémique n’est pas scientifique mais pratique. Ce n’est pas le virus qui est épidémique mais la cinétique de propagation et l’étendue des pathologies en résultant. Le SARS-CoV-2 est actuellement épidémique, comme l’est le coronavirus décimant les élevages porcins en causant la diarrhée ou alors les coronavirus détectés dans les élevages canins et causant eux aussi pas mal de dégâts.
2) Les coronavirus humains
Quatre coronavirus occasionnent des rhumes et quelques fois des infections dans la trachée ou les bronches sans occasionner de problème sanitaire important. Deux sont connus depuis les années 1960 et deux autres ont été identifiés dans les années 2000, ce qui ne signifie pas qu’ils soient apparus à cette date. C’est juste que les scientifiques ont porté une attention plus soutenue à ces virus depuis la crise épidémique causée par le SARS de 2003. Puis le MERS a été détecté en 2013 et enfin le SARS-2 en 2019. Les médias ont focalisé l’attention sur la protéine S et son domaine de liaison au récepteur ACE2, un domaine assez variable, pouvant muter et produire des variants censés être plus contagieux, voire capables d’échapper aux vaccins puisque ce domaine situé en position proéminente contient des motifs antigéniques (épitopes) déterminant la réponse humorale. En revanche, il n’y a pas de lien solide entre la virulence et la liaison ACE2. Le HCoV-MERS n’utilise pas le récepteur ACE2 et pourtant il est très méchant ; le HCoV-NL63 utilise le récepteur ACE2 et pourtant il n’est pas vraiment virulent. Pour connaître les causes virologiques du Covid-19, il faut chercher ailleurs
3 L’étrange protéine Spike
La protéine S est composée de deux sous-unités. La première contient le domaine de liaison (RND) pour ACE2 ; la seconde contient le domaine FP, situé près du second site de clivage. Ce domaine permet la fusion et l’entrée du virion, avec les autres domaines, les heptades, HR1, HR2, le domaine transmembranaire, TM et l’endo-domaine, E. Cette partie composite assure la translocation du virion dont le génome traverse le cytoplasme en direction de l’appareil ribosomal.
= NTD==RBD==SD1=SD2 = S1↓S2== ↓FP=HR1=HR2=TM=E=C-term
===== sous-unité S1 = /S1↓S2 / ==== ↓ S2’ === sous unité S2 =====
Le domaine de fusion FP est conservé sur la plupart des coronavirus. Le premier site de clivage S1/S2 est absent sur la protéine S du SARS-CoV. En revanche, la séquence situé sur la sous-unité S2 juste après le second clivage S2’ est conservée avec une précision remarquable. Elle contient le peptide de fusion FP.
4) Le code trypsine et les sarbecovirus
Les deux coronavirus ayant causé les syndromes pneumoniques aigus chez l’homme ont une origine animale. Les signatures protéiques en attestent. Je n’utilise pas les séquences génétiques pour étudier les similitudes. Une nouvelle tendance en virologie suggère de classer les virus à partir des familles de protéines. Je vais plus loin en cherchant des motifs protéiques conservés et traduisant les gains de fonction des virus. Des courtes séquences apparaissent déterminantes pour chaque étape fonctionnelle du virus et certains motifs ressemblent à des codes pour naviguer et opérer dans le tropisme intracellulaire. Examinons ces deux séquences de la protéine S récupérées sur un virus SARS-1 de 2003 et un SARS-2 de 2020 :
SARS-CoV-ZJ0301 ; GenBank : ABA02260.1 (2003)
781 __gfnfsqilpdplkptkr↓sfiedllfnkvtladagfmkqygeclgdinardlicaqkfngl
SARS-CoV-2 ; GenBank : QIG55857.1 (2020)
801 __nfsqilpdpskpskr↓sfiedllfnkvtladagfikqygdclgdiaardlicaqkfngltv
On observe une conservation parfaite d’un peptide de quelque 30 acides aminés au milieu duquel se trouve un motif polybasique (avec k et r). Il s’agit du peptide de fusion qui permet en quelque sorte au virion de s’accrocher au sas membranaire alors que le motif central est une sorte de code reconnu par la trypsine et qui permet au virion d’entrer dans la cellule. Ce code n’est pas le même. Le code lkptkr a été trouvé sur des virus infectant les chauves-souris ainsi qu’un bon nombre de variants extraits de civette palmée. Le code skprkrs est en revanche propre à un réservoir incluant les chauves-souris et les pangolins (trouvé sur des dizaines de génomes). Ces deux codes sont spécifiques des variants ou souches appartenant à la sous-famille des sarbecovirus (incluse dans la famille bêta) On ne les retrouve pas sur d’autres coronavirus, par exemple le A515 qui infecte les chauves-souris et appartient la famille alpha. Ce code skprkrs est présent sur la souche controversée RaTG13 récupérée en 2013 sur une chauve-souris mais séquencée en 2020. On le trouve aussi sur la souche Rc-0319 récupérée en 2013 au Japon et séquencée elle aussi en 2020.
Ces données penchent en faveur d’une origine animale du SARS-CoV-2 et plus exactement, un franchissement de la barrière inter-espèces du sous-clade pangolins et chauve-souris de 2013 (Le SARS-1 provient du sous-clade de 2002 civette et chauve-souris). Le classement phylogénétique des virus n’en reste pas moins approximatif et incertain, contrairement à la phylogenèse des espèces. Qui définit un clade en virologie ? Le génome, le réservoir ou alors les codes et fonctions ? Dans le cas des codes et des réservoirs, on peut assigner le SARS-1 au clade Sarb-2000 et le SARS-2 au clade Sarb-2010. Pour passer de l’un à l’autre on soustrait la civette, on ajoute le pangolin et on change le code trypsine. Des virologues français viennent d’annoncer la découverte de deux variants appartenant à ce réservoir et récupérés en 2010 au Cambodge sur des chauves-souris.
5) L’étrange code furine
La protéine S du SARS-CoV-2 a la particularité de disposer d’un code que l’on ne trouve pas sur le SARS de 2003. Ce code permet un clivage par la furine. Il est soupçonné d’avoir rendu fortement pathogène le coronavirus de 2019. Ce gain de fonction est un classique, observé sur d’autres virus. Le lien entre les codes polybasiques et la virulence des souches grippales est établi par des dizaines d’observations sans pour autant être une règle universelle. Les virus de la grippe aviaire hautement pathogènes avec les sous-types d’hémagglutinine (HA) H5 et H7 évoluent à partir de précurseurs faiblement pathogènes par l’acquisition de plusieurs résidus d’acides aminés basiques permettant le clivage de l’HA.
La séquence en question se présente ainsi :
671 casyqtqtnsprrar↓svasqsiiaytmslgaensvaysnnsiaiptnfti
C’est ce motif sprrars qui a nourri les thèses sur une origine artificielle du virus qui aurait été bricolé à partir d’un sarbecovirus de chauve-souris, voire de pangolin, en insérant une courte séquence nucléotidique permettant la production du code furine sur la protéine S. Le problème, c’est que le SARS-CoV-2 diverge de plus de 4% par rapport à la souche la plus proche, la RaTG13, et encore plus des autres souches appartenant au sous-clade Sarb-2010.
6) Nucléocapside
Si l’attention a été focalisée sur ma protéine S, la nucléocapside qui s’associe au génome est tout aussi importante. La protéine N de la nucléocapside possède plusieurs motifs polybasiques ainsi qu’un domaine riche en sérine, pouvant être phosphorylé ; et qui a une importance fonctionnelle précise. Voici la protéine N complète :
IDR — (RNA binding) — (SR rich LRK) — (RNA binding NLS) — IDR
1_____44__________182_____________247____________366___422
Et maintenant observons le domaine riche en sérine qui est extrêmement conservé, du SARS-1 au SARS-2 en passant par d’autres variants comme le Rc-o319 de la chauve-souris japonaise isolée en 2013. De part et d’autre du peptide enrichi en sérine nous voyons également une conservation très importante. Ces détails indiquent clairement la signature et la marque de fabrique du coronavirus de Wuhan responsable de la pandémie de Covid-19 :
SARS-CoV
152 pnnnaatvlqlpqgttlpkgfyaegsrggsqassrsssrsrgnsrnstpgssrgnsparmasggg
SARS-CoV-2
152 pannaaivlqlpqgttlpkgfyaegsrggsqassrsssrsrnssrnstpgssrgtsparmagnggd
Bat Rc-o319
152 pannaaivlqlpqgttlpkgfyaegsrggsqassrsssrsrgssrnttpgssrgnsparsvgngg
7) La protéine accessoire ORF6
Une troisième protéine mérite une attention, c’est la protéine dite accessoire ORF6. Que l’on ne confondra pas avec la protéine non structurale nsp6 qui elle, est codée sur la longue séquence de lecture ORF1a,b. Cette protéine codée par la séquence de lecture numérotée en 6 serait responsable d’une interférence avec la voie de signalisation JAK/STAT qui règle la balance entre immunité et inflammation. ORF6 produit une suractivation de STAT3 qui avec les cascades inflammatoires explique les formes graves du Covid. Cette petite protéine de quelque 60 résidus est elle aussi très conservée. On retrouve la signature du réservoir de 2013 d’où est issu le coronavirus de Wuhan, autrement dit le clade Sarb-2010. Cette séquence ORF6 est très conservée, y compris dans la souche japonaise, ce qui laisse penser que ORF6 n’est pas si accessoire que sa dénomination ne l’indique :
SARS-CoV-2 (2020)
mfhlvdfqvt iaeilliimr tfkvsiwnld yiinliiknl sksltenkys qldeeqpmei d
Rc-o319, GenBank : BCG66631.1 (2020)
mfhlvdfqit iaeilmmimr tfkvsilnld yiisvivrhl sksltenkys qldeeqpmet d
Pangolin GenBank : QIG55949.1 (2020)
mfhlvdfqvt iaeilliimr tfkvsiwnld yiinliiksl skpltenkys qldeeqpmei d
8) La signature S1 du SARS-CoV-2 et le code furine
Nous savons quelle est l’origine zoonotique du nouveau virus avec trois signatures permettant d’authentifier le clade, (i) sous-unité S2 et code trypsine, (ii) protéine N et domaine enrichi en sérine, (iii) protéine accessoire ORF6. D’autres signatures sont aisément identifiables. La virologie du SARS-CoV-2 s’est focalisée sur le site de clivage furine qui fait sa spécificité et ne se retrouve sur aucune souche du clade Sarb-2010, ni celui de 2000. La souche la plus proche est la RatG13. Nous pouvons superposer les résidus 660 à 720 et voir la similitude presque parfaite sauf l’insertion prra (un détail, la proline p est un acide aminé particulier capable d’interrompre des structures secondaires des protéines).
RaTG13
RaTG13 : ecdipigagicasyqtqtnsr↓svasqsiiaytmslgaensvaysnnsiaiptnftisvtt
SARS-2 : ecdipigagicasyqtqtnsprrar↓svasqsiiaytmslgaensvaysnnsiaiptnfti
J’ai par ailleurs fait une recherche sur trois motifs plus ou moins étendus montrant des similitudes (repérées à l’œil nu). Ces trois motifs situés vers 500-600 sont présents dans le SARS de Wuhan et la souche RaTG13 et en supplément, quelques autres souches de chauves-souris ainsi que de pangolins que je note ainsi :
580 tleilditpcsfggvsvitp : quatre « Bat » dont YN2018A ajouté en septembre 2019 et neuf pangolins ajoutés depuis décembre 2019, donc avant le séquençage de Wuhan
558 flpfqqfgrdiadttdavrdpqtleilditpcsfggvsvitp : ce motif plus large qui contient le précédent est présent sur quatre souches de pangolins
605 snqvavlyqdvnctev : cet autre motif se retrouve sur neuf souches de pangolins. Vous noterez qu’y figure l’acide aminé D en position 614, promis à devenir G lors de la mutation D614G enregistrée dans les séquences et devenue dominante sur la planète.
En revanche, ces motifs ne se retrouvent pas sur la souche Rc-o319 récupérée au Japon en 2013. Ce détail penche en faveur d’une transmission zoonotique du virus localisée en Chine.
9) Epilogue, d’où vient le SARS-CoV-2 ?
Nous connaissons le clade d’où provient le coronavirus pandémique de Wuhan et la localisation en Chine des animaux ayant pu contaminer les humains. Il reste néanmoins deux problèmes. D’abord la souche originelle, qui ne correspond à aucune des souches séquencées. La RaTG13 est la plus proche mais elle diverge de 4%, ce qui constitue tout de même 1200 nucléotides. Pour info, les variants du coronavirus pandémique ayant le plus muté affichent au compteur une divergence de 40. L’origine suppose un chainon manquant, que l’on nomme SARS-X, proche du RaTG13, mais avec cette insertion de quatre résidus constituant le code furine, ce qui représente le second problème. Il n’y a qu’une alternative pour expliquer ce schéma :
SARS-X → → gain de fonction ; code furine → → SARS-CoV-2
Première possibilité, la piste du laboratoire. Une souche SARS-X de corona aurait été utilisée pour réaliser une expérience ce gain de fonction, GOF, assortie ou non d’une insertion réalisée par génie génétique. Cette nouvelle souche serait sortie accidentellement du laboratoire, sans doute en contaminant un chercheur. Rappelons que le principe d’un labo P3 ou P4 est d’éviter de disséminer un agent pathogène dans la nature mais aussi de laisser sortir le chercheur pour qu’il retourne dans son foyer chaque jour.
Deuxième possibilité, le SARS-X infecte un animal, chauve-souris ou pangolin, et se transmet vers un humain, sorte de patient zéro, hôte pouvant réaliser une expérience de gain de fonction naturel assorti d’une insertion du code furine. En réalité, la Nature ne fait que produire des gains ou des pertes de fonction en modifiant les souches et variants de virus.
Les deux hypothèses sont plausibles mais ne sont pas équiprobables. La fabrication d’un virus est un événement concevable mais l’accident de laboratoire est très peu probable. La piste naturelle est inverse, le gain de fonction reste énigmatique mais une transmission de virus n’a rien d’improbable et d’ailleurs, elle a été observée avec le SARS de 2002-2004. Chacun se fera son opinion. Pour ma part, je crois à la piste naturelle en notant cependant que le gain de fonction acquis par le SARS-X place la science face à une grande interrogation. La sélection darwinienne de virus dont le génome mute n’explique pas comment ce gain de fonction est soudainement apparu dans un « patient zéro » pour générer le coronavirus pandémique. Nous sommes encore loin de la compréhension moléculaire du vivant. La crise sanitaire doublée de l’énigme virologique devrait inciter la science à mener un effort conceptuel et théorique sans précédent, quitte à casser les cadres épistémologiques hérités des années 1960.
10) Que peuvent faire les enquêteurs de l’OMS ?
Si l’on retrouvait l’hypothétique SARS-X sur un animal, la piste naturelle deviendrait privilégiée pour ne pas dire certaine. Quant à la piste du laboratoire, en admettant qu’elle tienne la route, on se doute bien qu’il est facile pour les responsables de détruire toutes les preuves en effaçant les cahiers de labo, qu’ils soient en support papier ou dans les mémoires d’ordinateur.
Bernard Dugué sur Agoravox